This article focuses on gaining approval for clinical research involving NHS patients, although the principles can be applied to other types of research. It can be quite a daunting process for the uninitiated applicant.
Often it can be made less drawn out by discussing the application with other colleagues in your department and looking over previous approved applications which very often have significant convergence with your own. This can save a lot of time in the initial phase and generally smooth the process. The intention is to give an overview of the requirements for setting up a research study, but it is not comprehensive. Specific arrangements for setting up studies vary within the UK and readers should refer to their own trust guidelines and regional policy documents. A list of references and websites is provided at the end of the document for further sources of guidance.
Ethical principles in research
The requirement for ethical approval exists in order to safeguard the ethical standards of practice in research. History has taught us that physicians don’t always uphold high ethical standards in the treatment of their patients [1,2]. The Nazi and Imperial Japanese medical experiments are among the most extreme examples of disregard for the ethical treatment of research subjects.
The Declaration of Helsinki is an important policy document of the World Medical Association (WMA) which sets out international standards for the ethical behaviour of physicians. All ethical applications follow the principle enshrined in this document-principles.
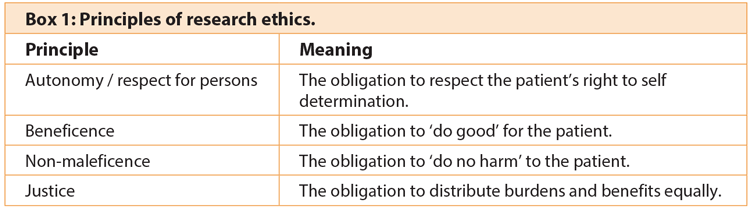
A commonly used ethical framework in healthcare (Box 1) identifies the principles of autonomy, beneficence, non-maleficence and justice [3]. These will be key concerns of any committee reviewing your application.
1. Autonomy / respect for persons:
The following rules apply to a person’s participation in a trial. A person should be:
- Properly informed (made aware of the risks, benefits and alternatives of the planned intervention)
- Not coerced into participating
- Free to withdraw at any time without needing to justify their decision
- Not penalised for refusal to participate
This principle recognises the autonomy of potential research subjects and the need to protect those with reduced autonomy. To be considered fully autonomous, a person must be competent to make the decision and be in a position to make the choice voluntarily. Certain institutionalised groups such as prisoners require safeguards to ensure they are protected from pressure to comply.
On the other hand, participation can provide benefits and individuals or groups should not be excluded on the basis of inconvenience or marginalised status.
2. Beneficence / non-maleficence
The purpose of beneficence is to ‘Maximise possible benefits and minimise possible harms’ [4]. This definition overlaps with the principle of non-maleficence or ‘do no harm’. It follows that the potential benefits to the individual should outweigh the risks. Even in the course of well-conducted research it cannot be guaranteed that no harm will be done, but robust precautions need to be in place to mitigate risk to the patient.
3. Justice
This principle focuses on the distribution of the burdens and benefits of research. It dictates that decisions made on the care of individuals must be fair and not superseded by the needs of a group or wider society, where the rights and well-being of vulnerable people are sacrificed for the benefit of others.
Research governance
The Research Governance Framework guides best practice for all research performed within the NHS and social care organisations in the UK. The Department of Health document Research Governance Framework for Health and Social Care [5] sets out guidelines in five main areas: ethics; science; information; health, safety and employment; finance and intellectual property (Box 2).
Box 2: Research Governance Framework principles of good practice
-
Ethics: Dictates that the primary concern in any research study is the well-being and rights of the participants. Integral to this is informed consent and data protection.
-
Science: Dictates that only high quality research should be performed. Research should be subject to peer review and should not be unnecessarily duplicated.
-
Information: Dictates that the findings of the research must be documented and accessible.
-
Health, safety and employment: Dictates that health and safety regulations are adhered to for the protection of participants and researchers.
-
Finance and intellectual property: Dictates that funding is appropriately consigned and authorship is correctly credited.
All researchers working with ‘human participants, their organs, tissue or personal data’ are required to understand and follow the principles of good practice set out in the Research Governance Framework.
1. Ethics
All research must have research ethics committee approval before it can begin. The committee requires evidence that appropriate arrangements be made to protect the “dignity, rights, safety and well-being” of the research participants. These measures include: obtaining informed consent, protecting participant data (Data Protection Act 1998), and compliance with the rules governing the storage and use of human tissue (Human Tissue Act 2004) where appropriate.
Researchers should consider the diversity of the population and involve patients and the public in the design and conduct of the study where possible.
2. Science
This part of the framework underlines the importance of high quality research. To ensure the scientific quality of the research, all proposals should be subject to scrutiny by independent peer review. Unnecessary duplication of work and poor quality research is considered unethical. Rigorous and precise record keeping must be maintained.
Trials of medicines and new devices are regulated by the Medicines and Healthcare products Regulatory Agency (MHRA) under the Medicines for Human Use (Clinical Trials) Regulations 2004 (Box 3). Research should follow the principles of good clinical practice (GCP).
Box 3: Clinical trials of investigational medicinal products (CTIMPs)
CTIMPs are studies in which the safety or efficacy of a new drug or device is being tested. Approval must be sought from the MHRA before the study begins. The MHRA is a government agency within the Department of Health. Application to MHRA is made through IRAS. CTIMP applications can only be reviewed by certain recognised RECs.
A clinical trial authorisation (CTA) must be issued by the MHRA before a trial can begin. The application can be made through IRAS, but a unique EudraCT number has to be obtained before the application is made. This can be done online via https://eudract.ema.europa.eu
Good clinical practive (GCP) training is a legal requirement for researchers involved in CTIMPs. GCP is “an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involved the participation of human subjects” (www.ich.org).
There are also recommendations within this section of the framework for retention and retrieval of data after the study ends.
3. Information
There should be access to research being conducted. The International Committee of Medical Journal Editors (ICMJE) requires that clinical trials are registered on a database that is accessible by the public. This can be done via www.isrctn.com or www.clinicaltrials.gov. It will not be possible to publish the clinical trial unless it is registered.
After scientific review, the research findings should be made available in a format that is understandable to the public.
4. Health, safety and employment
The safety of research participants, researchers and all other staff must be protected. The Health and Safety at Work Act 1974 must be observed.
5. Finance and intellectual property
This section covers guidelines on use of public funds in research, the protection of intellectual property rights and indemnification against harm to research participants.
Applying for ethics approval
The Health Research Authority (HRA) is an NHS organisation, launched initially as a special health a uthority in December 2011 to ‘promote and protect the interests of patients and the public in health research’. It was established to streamline the approvals process for health research. The aim is to establish it as a non-departmental public body in due course. The HRA now oversees the National Research Ethics Service (NRES), previously under the auspices of the National Patient Safety Agency (NPSA). The NRES will continue to function as the body managing ethical review for clinical research in the UK. The stated purpose of NRES is twofold:
- To protect the rights, safety, dignity and well-being of research participants.
- To facilitate and promote ethical research that is of potential benefit to participants, science and society.
These functions are implemented by a process of ethics review, performed by research ethics committees (RECs). Each REC consists of both lay members and people with particular expertise relevant to the application, including healthcare professionals and academics. A committee can have up to 18 members, of which at least a third are lay members [6]. Decisions within research ethics committees are generally reached by consensus rather than by majority vote [7]. The desired outcome is for the group as a whole to be comfortable with the final decision [8].
A typical applicant to NRES for ethics approval may be a healthcare professional, an academic or student, a pharmaceutical company or medical device company. If the project is university based and will not take place within the NHS or use NHS patients, local university ethics committees are used.
Ethics approval can’t be granted retrospectively, so must be sought before a study starts. Applications to RECs are made using the Integrated Research Application System (IRAS). The system was developed to improve the process of obtaining the various approvals required to conduct research in the NHS, of which ethical approval is one part (Box 4). The whole application is completed online including the patient information sheet (PIS).
Box 4: Using IRAS
-
IRAS is accessed online under www.myresearchproject.org.uk
-
An IRAS account is simple to set up with an email address and chosen password. There is no charge to set up an account.
-
Multiple projects can be entered within an account.
-
Resources within IRAS to guide researchers:
- Tabs that can be launched from the homepage: e-learning module, help page and ‘contact us’ (provides a telephone contact and email address for technical support as well as an email address for other queries and links to helpful websites).
- Question-specific guidance: green icons are attached to the questions within the application form. This guidance is particularly helpful when answering project filter questions.
-
When a new project is created, it opens to a navigation page. On the left side of the page is an access panel for all the forms that can be captured in IRAS.
-
The ‘Integrated Dataset’ (‘Full set of project data’): for each project, the information on the Integrated Dataset can be used to populate other forms within the IRAS system. This avoids the need to enter duplicate information for different approval forms.
-
Care is needed when answering questions in the project filters. IRAS is designed to generate further questions relevant to the answers given in the project filters. If the answer in the filter is not correct, the subsequent questions may not be appropriate for the study.
Requirements for a favourable REC opinion
The following is a guide to the issues that may be discussed during ethics committee deliberation. Any serious application would already encompass all these aspects anyway. They are reflected in the project data questions in IRAS [9,10].
1. Scientific design and conduct of the study
- Is the study design and methodology (including statistical aspects) sound and how has it been assessed?
- What assessment has been made of the risks versus burdens for the research participants?
- Are criteria for early subject withdrawal or study termination specified?
- Are provisions in place for monitoring the research?
- Is the research site equipped to fulfil its role? Are there sufficient numbers of staff with appropriate expertise, are the facilities adequate and is there an infrastructure in place that can support the study?
- What plans are in place for reporting and disseminating the results of the research?
Therefore it is vital to have the study appropriately costed and resourced at the outset, with database and information systems in place as well as statistical support and power calculations to support subject / sample numbers required.
2. Recruitment of research participants
- Can the choice of population from which the subjects will be recruited be justified, to ensure that no group is unfairly overburdened or denied an opportunity to participate?
- What are the inclusion and exclusion criteria?
- How will research participants be approached and what methods will be used for recruitment?
- What information will be provided to research participants? (Guidance on preparation of the patient information sheet (PIS) can be
found on the NRES website.)
3. Care and protection of research participants
- What are the risks of any proposed intervention?
- Will standard therapies be withheld from research participants and can this be justified?
- What care will be provided to the patient during the study and after it ends?
- Will costs to patients be reimbursed?
- Are the qualifications and expertise of staff suitable for their role in the study?
- What insurance and indemnity arrangements are in place in the event of harm to a research participant?
4. Confidentiality
- Who will have access to the data?
- What measures will be in place to protect identifiable data?
- What long-term arrangements are in place to store the data?
5. Informed consent
- Consent process (guidance on preparation of the consent form can be found on the NRES website).
- How and by whom will subjects be approached?
- Is the autonomy of the subject recognised?
6. Community considerations
- What is the relevance of the research to the population from which participants are recruited?
Arranging REC review
At present, for studies involving NHS patients or NHS property, formal application must be made to a REC through NRES. The routes for application are via the Central Allocation System (CAS) and Local Allocation system (LAS).
An application may also be made directly to a specific REC. Questions for completion of the REC form (completed within IRAS) are in line with the best practice guidelines of the Research Governance Framework, as described above.
Contact telephone numbers for CAS, LAS and local RECs are available through the NRES website. The operator ensures the appropriate allocation system is being used and may direct the caller to the alternative service after a series of questions.
At the time of writing, research under the following categories must be allocated via CAS:
- Clinical trials of investigational medicinal products (CTIMPs)
- Research of medical devices
- Research involving prisoners
- Research involving adults lacking capacity
- Establishing research tissue banks
- Projects funded by the US Department of Health and Human Services (DHHS)
- Establishing research databases
After the booking is finalised, a REC reference number is allocated. A document is then sent out confirming the meeting venue and date, contact details of the local REC co-ordinator and paperwork requirements. The REC co-ordinator can be a helpful contact point for any queries or concerns prior to the REC committee meeting.
All the relevant paperwork should be ready at the time of booking, as a full hard copy application to the REC must be submitted within four working days. Failure to carefully follow the instructions specified after booking may void the application.
The NHS REC form is populated from the Integrated Dataset in IRAS and can be printed for submission to REC. The correct REC name and reference number should be added to the application form with the same lock code on each page.
All required authorisations must be made before the application is submitted. Most signatures can be performed electronically using the IRAS system. However, it is important to be aware that any changes made after electronic authorisation can invalidate the form. The application must therefore be complete and ready for submission before signing off. (NB: the ‘proceed to submission’ button for each form on the IRAS system does not submit the form to any organisation. Selecting this button stores the form in the submission history with a code and this can be printed out at any time with the code on each page.)
The REC also requires copies of supporting documents (with a version number and date). These include the research protocol, patient information sheet and patient consent form.
Although it is not compulsory to attend the REC meeting, it allows committee members to ask any questions that arise during the meeting. This may help them reach a decision in a shorter timeframe. Therefore, it is strongly advised you attend this. Most questions finally centre upon how the study is explained in the PIS and consent forms. It is useful to seek the opinion of lay friends and family as they often can give you proper insight into whether forms are clear and intelligible. Also remember not to overburden the document with complex terminology.
The REC is required to provide an opinion within 60 days of receipt of a valid application. If the opinion of the REC is a request for further information, the clock stops. After the meeting, the committee can request clarification from the applicant only once. If an unsatisfactory response is received, the committee may give an unfavourable opinion or allow the applicant to resubmit a modified response.
Proportionate review
Proportionate review has been introduced to speed up processing of applications that are thought to pose low risks to participants. Guidelines for eligibility are available through the NRES website, using the No Material Ethical Issue Tool (NMEIT).
Sponsorship
Sponsorship is required for all clinical research studies. Proof of sponsorship is necessary to start a study and provides assurances that appropriate arrangements have been put in place for running the project. The funding source may not be the same as the sponsor.
The role of the sponsor is defined by the UK Clinical Trial Regulations as “an individual, company, institution or organisation which takes responsibility for the initiation, management and / or financing of a clinical trial”. In practice it would be unusual for an individual to take on the full financial and legal burden of sponsorship.
For studies within the NHS, the sponsor may be a commercial company, the NHS Trust or the university with which a clinical academic holds a contract.
After ethics approval
The Research Governance Framework stipulates that any research taking place within an NHS organisation must be approved by the host NHS organisation before it starts. This is required in addition to receipt of a favourable opinion from a REC.
Applications for approval (NHS permission) are made to the Research Management and Governance (RM & G) Department within the host organisation. This is also known as Research and Development (R&D) approval.
In contrast to the ethics approval process, where REC approval is transferable throughout the UK, NHS permission is required separately from each site to conduct a study. A Site-Specific Information (SSI) form must be generated through IRAS for each site involved and reviewed by the NHS R&D office for that site. Each NHS trust and primary care trust has its own internal arrangements for approving research within their organisation. At present there is no strict timeline to achieve NHS permission. It is particularly crucial to be aware of this if attempting to set up a multicentre study, as it can be an important source of delay [11-14].
A number of issues can be raised at this stage including the true costs of the study, equipment requirements and utilisation of NHS resources. Do not expect the R&D office to be proactive in chasing up their concerns or you may find that your application is at a standstill when you had assumed it was in process. Clarify exactly the requirements for each R&D office and deal with them promptly otherwise your study risks being held in a no-man’s land of bureaucratic stagnation. Remain prepared for additional unexpected hurdles that arise during the approval process.
Approval barriers may be relatively easy to resolve e.g. reformatting a document or seeking an additional confirmation from the department head that additional costs will not be incurred or are within the current NHS charges e.g. blood sample collection as part of routine care.
Other requirements for approval depend upon the study. The IRAS project data can be used to populate applications for some regulatory bodies including MHRA, Administration of Radioactive Substances Advisory Committee (ARSAC) and Gene Therapy Advisory Committee (GTAC).
National Institute for Health Research Coordinated System for gaining NHS Permission (NIHR CSP)
The Clinical Research Network (CRN) is part of the National Institute for Health Research, and supports high quality research studies through conception to delivery.
Its remit includes the streamlining of gaining NHS permission. To be eligible for support from CRN, a study must be ‘adopted’ by the NIHR CRN Portfolio. Researchers in England need to complete a Portfolio Adoption Form (PAF) via IRAS for the study to be considered for adoption.
Conclusion
It is proper that physicians are expected to conduct their research to high ethical standards and safeguards must be in place to ensure governance in clinical research. However, the processes involved in achieving various regulatory approvals can be complex, confusing and longwinded. These difficulties are recognised and there are ongoing efforts to streamline approval applications and improve support so that good clinical research can continue.
It is important not to underestimate the amount of preparatory work and time that may be required to set up a clinical research project. Without ethical approval the study can’t begin, but it is only one part of a complicated and sometimes heavily bureaucratic process. Predicting potential hurdles and seeking advice from experienced colleagues is strongly recommended.
References
1. Emanuel EJ, Crouch RA, Grady C, et al. The Oxford Textbook of Research Ethics. Oxford University Press; Oxford, UK; 2008.
2. Fischer BA. A summary of important documents in the field of research ethics. Schizophrenia Bulletin 2006;32(1):69-80.
3. Beauchamp TL, Childress JF. Principles of Biomedical Ethics First edition. Oxford University Press; New York, USA; 1979.
4. National Commission for the Protection of Human Subjects of Biomedical and Behavioural Research. Ethical principles and guidelines for the protection of human subjects of research (The Belmont Report). 1979.
5. Research Governance Framework for Health and Social Care: Second edition. Department of Health; UK; published online 24 April 2005.
6. Governance arrangements for research ethics committees: a harmonised edition. UK Health Departments; 9 May 2011 (Updated April 2012).
7. Moreno JD. Ethics by committee: the moral authority of consensus. Journal of medicine and philosophy 1988;13(4):411-32.
8. Research Ethics Committees. Basic concepts for capacity building. World Health Organisation; 2009.
9. Governance arrangements for NHS research ethics committees. Department of Health; UK; 2001.
10. Standards and Operational Guidance for Ethics Review of health-Related Research with Human Participants. World Health Organisation; 2011.
11. Rees M, Wells F. Falling Research in the NHS. A clear national strategy is needed to overcome local barriers to research. BMJ 2010;340: Editorials.
12. Snooks H, Hutchings H, Seagrive A, et al. Bureaucracy stifles medical research in Britain: a tale of three trials. BMC Medical Research Methodology 2012;12:122.
13. Thompson AGH, France EF. One stop or full stop? The continuing challenges for researchers despite the new streamlined NHS research governance process. BMC Health Services Research 2010;10:124.
14. Al-Shahi R. Research ethics committees in the UK- the pressure is now on research and development departments. J R Soc Med 2005;98:444-7.
Online resources for researchers
1. Clinical Research Network:
www.crncc.nihr.ac.uk
2. Clinical Trials Toolkit:
www.ct-toolkit.ac.uk
3. Data and Tissues Toolkit:
www.dt-toolkit.ac.uk
4. Experimental Medicine Toolkit:
www.em-toolkit.ac.uk
5. Ethics Research Information Catalogue:
www.eric-on-line.co.uk
6. Health Research Authority:
www.hra.nhs.uk
7. Health and Safety Executive:
www.hse.gov.uk
8. Integrated Research Application System:
www.myresearchproject.org.uk
9. UK Legislation:
www.legislation.gov.uk
10. Medicines and Healthcare products Regulatory Agency:
www.mhra.gov.uk
11. Medical Research Council:
www.mrc.ac.uk
12. National Institute for Health Research:
www.nihr.ac.uk
13. National Research Ethics Service:
www.nres.nhs.uk
14. UK Clinical Research Collaboration:
www.ukcrc.org
15. World Medical Association:
www.wma.net
Online resources for patients
www.invo.org.uk
www.rdslondon.co.uk
www.peopleinresearch.org
www.healthtalkonline.org/medical_research/
www.ucl.ac.uk/public-engagement
Declaration of competing interests:
Authors’ funding sources: UCLH Biomedicine NIHR, BHF, MRC, Wellcome Trust and Boston Scientific Research Fellowship (Vanessa Cobb).



