Research is the process of acquiring new generalisable knowledge and should be fully integrated into healthcare work. There is a growing drive to encourage and further develop evidence-based practice in medicine so that staff and patients benefit from improved healthcare provision and clinical outcomes. It is a mandatory requirement that all clinical research studies should receive a letter of NHS permission prior to starting (also referred to as R&D approval). This article comes as an introduction to research governance requirements in healthcare and will describe the various considerations in setting up a research study and gaining NHS permission.
The Research Governance Framework (RGF) [1] was issued in 2001 to outline the principles of governance that apply to all healthcare research. Research governance is a key standard for healthcare organisations and includes the Research Ethics Committee, Data Protection Act, Mental Capacity Act, Human Tissue Act, Ionising Radiation Regulations and Medicines for Human Use Regulations. The main aim is to protect the dignity, rights, safety and wellbeing of participants as well as protect researchers by providing a framework to work within.
Getting started
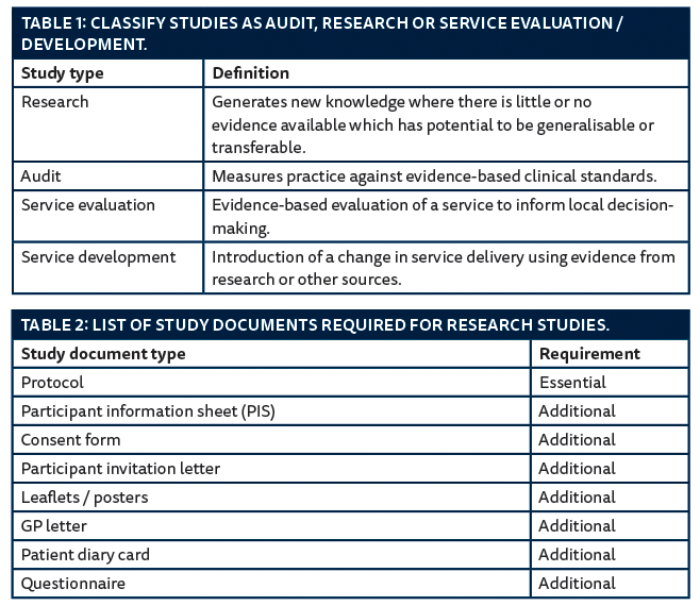
For those working in healthcare, research ideas tend to be driven by clinical need. The usual time and financial constraints will need to be addressed but the benefits tend to outweigh the dedication and time commitment required. Following the initial idea, consideration needs to be given to whether the proposed study is research, audit or service evaluation [2,3] (Table 1).
Once the study has been classed as research the next step would be to contact the local Research and Development (R&D) department at the local trust or university. R&D will be able to guide researchers through the various processes of research governance. Consideration will need to be given to the scale of the project as this will have an effect on the timelines, resources and staff commitment.
The chief investigator is the individual who takes overall responsibility for the design, conduct and reporting of the study. The principal investigator is responsible for the conduct of research at a specific study site. Table 2 lists common types of study documents required for conducting a research study. The National Research Ethics Service (NRES) has templates for the participant information sheets and consent forms that can be adapted to the needs of the study [4].
The local R&D may be able to offer further advice on protocol templates and available resources such as running a literature review and data analysis. The protocol should demonstrate sound background information on the scientific justification of the proposed study, appropriate study design and methodology to include participant identification, recruitment and consent, as well as outline statistical aspects such as number of recruits and statistical analysis. Patient and public involvement (PPI) is becoming increasingly important and several trusts and universities already have available panels that researchers can access [5].
Once the study documents have been finalised the researcher may need to obtain evidence that the project has received favourable independent scientific review. If the study has secured external funding this process is usually undertaken by the funding body at the time of the funding application. For unfunded or student studies, check with the local R&D or university for the specific scientific review requirements. Evidence of scientific review is required by the ethics committee reviewing the study.
Depending on the type of study, research costs need to be considered at an early stage as identified from the protocol, and funded accordingly. Local R&D support may be available to help researchers identify relevant funding bodies that will be suitable for urology-related studies. Preparing grant applications for local or national funding can be time-consuming so allow plenty of time to prepare the relevant paperwork to include collaborative links, study costs and any pilot data that may support the application. Furthermore, many of the larger funding bodies (such as the National Institute for Health Research) now require applications to have had Patient and Public Involvement in the design of the research. R&D will require a copy of the funding letter or other relevant supporting information to ensure that research-related costs are attributed and resourced appropriately.
Typical research costs to be considered in the setting up of the study include study staff time, administrative fees including printing and postage, pharmacy, laboratories, radiology, local fees, etc. Study activity that is delivered as part of standard care does not have to be included in the costs, however, it is important when describing the study that it is indicated clearly which activities will be part of standard clinical practice, and which activities are research-specific.
Portfolio adopted studies consist of high-quality studies that benefit from infrastructure and financial support from the National Institute for Health Research. The network supports research nurses, support staff, data collection and use of facilities, tests, or services where research activity adds value to patient care. Directorates receive money per patient recruited, and portfolio activity informs allocation of ‘strategic’ research funding to NHS trusts. Portfolio and commercial studies are performance managed, so it is essential that studies are initiated quickly and patients are recruited to time and target.
Research governance requires that all health-related research has a formal sponsor. The sponsor is the organisation with responsibility for securing arrangements to initiate, manage and finance the study, e.g. an NHS trust, a university or a commercial company. A sponsor representative will be required to authorise various application forms, e.g. for NHS ethics applications, and research contracts and agreements. The sponsor will negotiate contracts and agreements with relevant parties and will also look into any arising intellectual property that needs to be considered. Commercial research follows the RGF principles and the company funding the study is the sponsor responsible for the conduct of the study.
Ethical considerations
Prior to gaining NHS permission, consideration needs to be given to the ethical principles of the proposed study. The NRES comprises the Research Ethic Committees (RECs), aiming to protect and promote the interests of patients and the public in health and social care research [6]. RECs review applications for research and give an opinion about the proposed participant involvement and whether the conduct of the proposed research is ethical. Submission is via the online Integrated Research Application System (IRAS) [7]. The type of ethical review required needs to be confirmed with local R&D and it depends on the type of study to be conducted. NRES have a useful decision tool available to researchers via their website [8]. For studies involving anonymised previously collected clinical data or tissue as part of standard care no NRES approval is required but the study still needs NHS permission. Most universities have got their own ethical committees for student and staff projects that do not require NRES review. Typical studies requiring review by the NRES are listed below:
- Patients and users of the NHS.
- Individuals identified as potential research participants because of their status as relatives or carers of patients and users of the NHS.
- Access to data, organs or other bodily material of past and present NHS patients.
- The recently dead in NHS premises.
- The use of, or potential access to, NHS premises or facilities.
- NHS staff – recruited as research participants by virtue of their professional role.
Applications are normally reviewed by a full REC and an opinion is received within 60 days. Where a research study presents ‘no material ethical issues’ it can be reviewed and approved by a proportionate sub-committee and the application is reviewed within 10 days of receipt [9].
Legislation acts and regulations
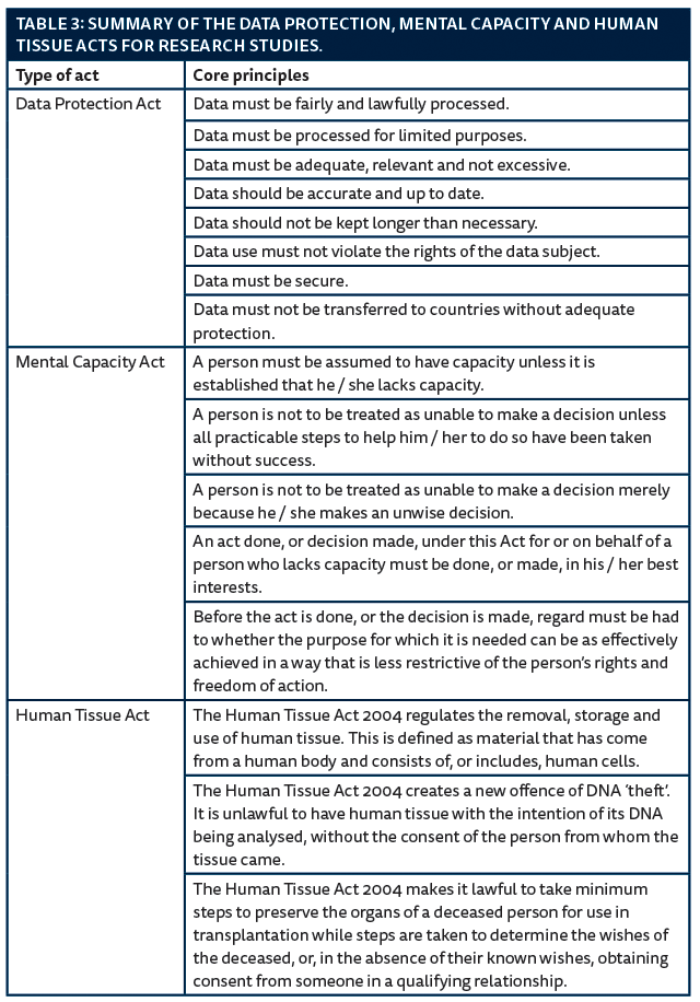
Three legislation acts form part of the RGF concerning data protection, mental capacity and human tissue. The three acts are only applicable in England, Wales and Northern Ireland as Scotland has its own legislation, summarised in Table 3. All clinical staff, as well as researchers, must comply with the Data Protection Act [10]. Typical issues to be considered in relation to data include storage, access, transfer, use and identification. Research data should, where possible, be anonymised (i.e. information that could be used to identify a person is removed) before being saved and / or transferred.
The Mental Capacity Act provides a comprehensive framework for decision-making on behalf of adults aged 16 and over who are unable to make decisions for themselves [11]. A person’s loss of capacity may be temporary, and capacity may fluctuate so that some people may lack capacity to make a complex decision but retain the capacity to make other decisions. Assessing capacity encompasses the ability to understand the information relevant to the decision, retain the information, use or weigh the information and communicate that decision by any means.
The Human Tissue Act makes consent the fundamental principle underpinning the removal, storage, use and disposal of human tissue [12]. Human tissue can be taken, stored and used for specific research, provided the project has REC approval. Consent is not required for research on tissue from living patients if the samples are anonymised and the project has REC approval; or if the tissue samples were obtained before 1 September 2006 (when the Human Tissue Act came into force).
The Ionising Radiation Regulations govern the exposure of research participants to ionising radiation. Typical procedures involving ionising radiation include x-rays and CT scans. MRI or ultrasound investigations do not involve ionising radiation. Where research involves the administration of radioactive substances, an Administration of Radioactive Substances Advisory Committee (ARSAC) certificate must be held at each research site where administrations take place. The local R&D will be able to support with relevant approvals.
MHRA
The Medicines and Healthcare products Regulatory Agency (MHRA) is responsible for regulating all medicines and medical devices in the UK. A Clinical Trial Authorisation (CTA) is required for any clinical trial of an investigational medicinal product (CTIMP). An application to MHRA devices will be required where the study is a clinical investigation of a medical device undertaken by the manufacturer for CE marking purposes. MHRA application may also be required for an investigation of a non-CE marked product, or an investigation of a CE marked product that has been modified or is to be used outside its intended purpose. MHRA approval is not always required in the case of medical devices manufactured ‘in-house’ in a healthcare establishment or clinician led off-label use of a medical device.
In such cases contact MHRA directly to discuss the purpose of the investigation and determine whether MHRA approval is required. For studies concerning investigational medicinal products (such as drugs) or devices, it is advisable to seek advice from the local R&D as soon as possible. General guidance on whether MHRA approval is required can be found on the MHRA website [13].
Contractual requirements
For those researchers who do not have a substantive contract with the NHS organisation but whose research involves the NHS, an honorary contract or a letter of access with the relevant NHS organisation is required. By issuing honorary contracts, the NHS organisation ensures that all researchers are contractually bound to take proper account of the NHS duty of care. Researchers with honorary contracts with the NHS are covered by NHS indemnity.
The research team should have up to date curriculum vitaes and, for the conduct of clinical trials, have current Clinical Good Practice (GCP) training. GCP is the standard and guidelines to which all clinical research is conducted. GCP training is a regulatory necessity for investigators in drug and device clinical trials.
Other considerations
It is advisable to maintain a study site file with all essential documents. After NHS permission, R&D should be kept informed of the study status and receive copies of annual reports, recruitment and study closure. For any changes to the study documents an amendment must be submitted to R&D for review and approval prior to implementation. Amendments are classed as substantial or non-substantial and only substantial amendments require ethical review. Investigators should arrange appropriate archiving of study files and information, usually for five to ten years after the study end. The results of the study should be published or presented to inform fellow colleagues and clinical practice.
Summary
In brief, the RGF sets out the principles required for the conduct of research in NHS healthcare settings. This article serves as an introduction to those principles and hopes to summarise the regulatory processes and approvals involved. Good research practice is underpinned by evidence-based practice and supports the quality of scientific evidence needed to drive improvements in patient healthcare. Adherence to research governance promotes patient confidence and study participation, as well as encourages researchers to demonstrate high standards in study conduct and integrity. NHS organisations have a duty to ensure roles and responsibilities are clear and that appropriate resources and skills are in place to deliver appropriate research. The RGF aims to enhance ethical and scientific quality, minimise risks and ensure adequate monitoring.
References
1. Department of Health. Research Governance Framework for Health and Social care: second edition. 2005.
https://www.gov.uk/
government/publications/
research-governance-framework
-for-health-and-social-care-second-edition
Accessed January 2014.
2. NHS Health Research Authority. Determine whether your study is research.
http://www.hra.nhs.uk/
applications/is-your-project-research/
Accessed January 2014.
3. Healthcare Quality Improvement Partnership. A Guide for Clinical Audit, Research and Service Review.
http://www.hqip.org.uk/assets/
Downloads/Audit-Research
-Service-Evaluation.pdf
Accessed January 2014.
4. NHS Health Research Authority. Information sheet and consent form guidance.
http://www.nres.hra.uk/
applications/guidance/
?EntryId62=67013
Accessed January 2014.
5. INVOLVE.
www.invo.org.uk
Accessed January 2014.
6. Department of Health. Governance arrangements for research ethics committees: a harmonised edition. 2011.
7. Integrated Research Application System.
www.myresearchproject.org.uk
Accessed January 2014.
8. NHS Health Research Authority. Medical Research Council.
http://www.hra-decisiontools.org.uk/ethics/
Accessed January 2014.
9. NHS Health Research Authority. Proportionate Review Service.
http://www.hra.nhs.uk/about
-the-national-research-ethics-service/
building-on-improvement/
proportionate-review/
Accessed January 2014.
10. Legislation.gov.uk. Data Protection Act 1998.
http://www.legislation.gov.uk/
ukpga/1998/29
Accessed January 2014.
11. Legislation.gov.uk. Mental Capacity Act 2005.
http://www.legislation.gov.uk/
ukpga/2005/9/enacted
Accessed January 2014.
12. Legislation.gov.uk. Human Tissue Act 2004.
http://www.legislation.gov.uk/
ukpga/2004/30/contents
Accessed January 2014.
13. Medicines and Healthcare Products Regulatory Agency.
http://www.mhra.gov.uk
Accessed January 2014.
TAKE HOME MESSAGE
-
The Research Governance Framework is a key standard research undertaken in healthcare organisations.
-
It is a mandatory requirement that all clinical research studies should receive a letter of NHS permission prior to starting.
-
Research should be embedded into clinical practice.
-
Support is available from local R&D offices who can advise on local research requirements.
Acknowledgements
The author would like to thank Dr Jim Lithgow, Clinical Research Coordinator at Sheffield Teaching Hospitals NHS FT, for his support in preparing this document.
Declaration of competing interests: None declared.